
Enfermedad
genética
Para esclarecer qué se entiende por
enfermedad genética, llamadas también hereditarias o congénitas, debemos
revisar el concepto de gen o partícula que trasmite caracteres de padres a
hijos.
Los genes están en los cromosomas,
estructuras filamentosas que se encuentran en el núcleo de todas las células
del cuerpo. Cada cromosoma tiene miles de genes diferentes.
Los genes se encuentran siempre en pares.
El padre y la madre proveen cada uno un gen de sus pares para formar un nuevo
par para el niño. De esta manera, los genes siempre pasan características
familiares de una generación a la siguiente.
Los padres no pueden controlar los genes
que heredaran sus hijos.
Así existen enfermedades de los padres cuya
información está contenida en genes y estos son capaces de traspasarse a los
hijos cuando se realiza la fecundación del óvulo materno.
Las enfermedades genéticas corresponden a
un grupo heterogéneo de afecciones que en su desarrollo presentan un
significativo componente genético. Ello puede ser alguna alteración en un solo
gen, en varios genes (poligenes) o en muchos genes (cromosomas).
La alteración genética puede producir
directamente la enfermedad (por ejemplo, el caso de la Hemofilia ) o
interactuar con factores ambientales (como, por ejemplo, la predisposición
genética en el desarrollo de la Hipertensión arterial).
Cada vez se hace más difícil separar las
afecciones de desarrollo ambiental de aquellas llamadas "genéticas puras".
A modo de ejemplo, conviene recordar que para varias enfermedades típicamente
ambientales, como infecciones bacterianas, parasitarias, etc., recientemente se
ha demostrado una susceptibilidad genética del individuo.
En síntesis, se puede decir que las
enfermedades genéticas son aquellas debidas a una alteración en el material
genético de una persona y que suelen provocar una alteración metabólica.
Se pueden distinguir:
Enfermedad genética dominante: enfermedad
genética en la que con una sola copia afectada del gen se manifiesta la
enfermedad.
Enfermedad genética recesiva: enfermedad
genética en la que se necesitan dos copias afectadas del gen para padecer la
enfermedad.
Enfermedad ligada al sexo: enfermedad que
se trasmite en los cromosomas sexuales; suele ser más frecuente que se trasmita
en el cromosoma X.
Enfermedad monogénica: enfermedad genética
debida a la alteración de un solo gen.
Enfermedad poligénica: enfermedad genética
debida a la alteración de diversos genes.
Algunas
enfermedades genéticas son: Sindrome de Down, Sindrome de Klinefelter,
Sindrome de Turner, Anemia Falciforme, Hemofilia, Daltonismo, Albinismo,
Polidactilia.









Síndrome
de Down
El Síndrome de Down es una alteración
cromosómica causada por la presencia de un cromosoma 21 de más. La alteración
comporta siempre unos rasgos morfológicos característicos y una disminución de
la capacidad mental, aunque la manifestación de estas afectaciones varía según
los individuos.
No existen, de todas formas, grados de
afectación del síndrome. Simplemente se tiene o no. Todas las personas tienen
46 cromosomas en cada célula de su cuerpo. En los casos de una persona con el
SD, existe un cromosoma de más en la pareja 21. Esta persona, pues, tiene 47
cromosomas en lugar de 46, de ahí la denominación de trisomía 21.
Se desconoce el origen exacto de esta
alteración, si bien es posible determinar la coincidencia de varios factores de
riesgo, tales como la edad de la madre.
El Síndrome de Down se puede diagnosticar desde
el nacimiento mediante una prueba genética denominada cariotipo.
Actualmente es posible realizar dos pruebas
de diagnóstico en el periodo prenatal, mediante una biopsia de corion (a las
10-13 semanas de gestación) o la amniocentesis (a las 15-17 semanas).
Espontáneamente uno de cada 600-700 recién
nacidos puede tener el Síndrome de Down, pero la influencia del diagnóstico
prenatal reduce esa cifra.
No existe ningún tratamiento médico ni
farmacológico para el Síndrome de Down. Hasta el momento el único tratamiento
consiste en la intervención educativa, que durante la primera infancia recibe
el nombre de atención precoz. Como primer eslabón de la integración social de
la persona con Síndrome de Down, siempre se debe acompañar de un entorno
familiar favorable.
http://www.terapiafisicasanisidro.com/images/sindrome%20de%20down.jpg
Síndrome
de Turner
Es una alteración cromosómica encuadrada
dentro de las disgenesias gonadales que cursa con talla baja, infantilismo sexual
y malformaciones somáticas.
Generalidades y Clínica
La constitución cromosómica 45 XO se da en uno
de cada 2.500 nacidos vivos con fenotipo femenino. Esta incidencia puede ser
mayor si se tiene en cuenta que el 5% de abortos espontáneos tienen este mismo
cariotipo.
Aun cuando su etiopatogenia no es bien
conocida, parece ser el resultado de una no disyunción materna o paterna. Se
han encontrado además diferentes mosaicos, delecciones del brazo largo del
cromosoma X e isocromosomas.
Estas pacientes presentan genitales externos
femeninos, con útero y trompas normales, pero de tipo infantil. No existen ovarios,
que están sustituidos por unas formaciones de aspecto acintado y color
blanquecino, situados en los ligamentos anchos. Microscópicamente se comprueba
que estas estructuras son de tejido conjuntivo fibroso, dispuestas en remolinos
y carentes de células germinales.
En los síntomas destacan tres aspectos:
1.- Talla baja
Suelen alcanzar en la edad adulta una talla
que oscila entre 129 y 147 cm según las diferentes series de la literatura.
Las etapas del crecimiento pueden sistematizarse
en cuatro fundamentales:
- Crecimiento intrauterino retrasado.
- Crecimiento normal hasta los 2 años.
- Progresivo deterioro del crecimiento entre
los 2 y los 11 años.
- Crecimiento lento en la adolescencia sin
estirón puberal.
Hay que tener en cuenta que existe una cierta
displasia esquelética en algunos casos.
2.-Infantilismo sexual
La disfunción más frecuente en un órgano en
el síndrome de Turner es la disgenesia ovárica, ya que el feto a las 14-16 semanas
de la gestación presenta ovarios de estructura histológica normal, así como
también lo son las trompas, el útero y la vagina. Es en el último trimestre del
embarazo y en los meses siguientes a la gestación cuando se produce una rápida
atresia de los ovocitos y fibrosis del estroma. Sin embargo, este proceso a
veces no es absoluto, y ello explica el que en un 5% de las enfermas puede
haber un grado de función ovárica residual suficiente para iniciar el
desarrollo de las mamas en la pubertad y algunas pueden tener una o más
menstruaciones espontáneas. Raramente tienen ovulaciones, habiéndose observado
casos de embarazos, tanto en niñas con monosomía como con mosaicos.
Las gonadotropinas permanecen en niveles
altos durante los primeros años de la vida (sobre todo la FSH), para luego
disminuir a niveles similares a las niñas normales. Al no producirse la
pubertad, por fracaso ovárico, aumentan las gonadotropinas hasta niveles de
menopáusicas, y por tanto no se produce el desarrollo de los caracteres sexuales
secundarios, y las afectas permanecen sexualmente infantiles y con amenorrea
primaria.
Hasta hace poco la infertilidad era
definitiva, pero gracias a las investigaciones en la donación de óvulos y su
implantación en el útero de la receptora, la maternidad es posible, aunque
deben resolverse algunos problemas como sincronizar los ciclos hormonales de la
donante y de la receptora, así como encontrar el momento óptimo para el trasplante
de embriones al útero.
Otro tema debatido durante años era el de la
extirpación de las cintillas ováricas. No hay ninguna razón para realizarlo, a
no ser que presenten signos de virilización o haya en el cariotipo un cromosoma
Y (muy raro), en cuyo caso hay riesgo de formación de gonadoblastomas.
3.- Anomalías somáticas
Son variadas y dependen muchas veces de los
diversos cariotipos.
En el momento del parto suele ser frecuente
el linfedema de las extremidades o el higroma de la nuca, ambos condicionados
por una hipoplasia de los vasos linfáticos superficiales (25% de los casos). Se
produce un bloqueo linfático, que en general desaparece durante el primer año
de la vida, aunque a lo largo de ella puede dar lugar a fenómenos de edema
intermitente y tendencia a la linfangitis.
El descenso de la línea de implantación
posterior del cabello y la rotación de las orejas, pueden ser consecuencia de
la distensión de la nuca.
Con frecuencia presentan grados variables de
disminución de la mandíbula (micrognatia) y secundariamente deficiente
implantación dentaria. Puede haber paladar ojival, epicanto, sinofridida y
disminución de la distancia interorbitaria.
El cuello presenta evidente acortamiento,
siendo más ancho de lo habitual. En una cuarta parte de los casos hay pterigium
colli. Hay una elevada frecuencia de esta anomalía en la monosomía sexual (45
X0). Este cuello alado se forma por unos pliegues cutáneos a modo de membranas,
de forma triangular que van desde la región mastoidea hasta el acromión (cuello
de esfinge).
El tórax es ancho, con aumento de la
distancia entre las mamilas (tórax en escudo) y no hay desarrollo en los casos
típicos.
Existen alteraciones óseas como el genu
valgo, escoliosis y otras anomalías ortopédicas. De las más frecuentes son el
cúbito valgo y el acortamiento del cuarto metacarpiano (73% y 54% de nuestra
serie respectivamente). Se ha descrito el signo del carpo ¾mucho más
inconstante¾ que es la disminución del ángulo formado por la extremidad
inferior del cúbito y la primera fila de huesos del carpo.
Suele haber osteoporosis.
Las anomalías cardiovasculares son
relativamente frecuentes, sobre todo la coartación aórtica (10%), la estenosis
aórtica y las valvulopatías (tricúspide y aórtica), aunque también pueden haber
defectos del tabique interventricular, malformaciones de los grandes vasos y
dextrocardia. Cada vez hay más descripciones de aneurismas disecantes de aorta,
de cuyo origen pueden ser responsables trastornos hemodinámicos o defectos
mesenquimales primarios.
Fuentes web:
http://serbal.pntic.mec.es/~jpons4/pag1.htm
www.profesorenlinea.cl - Registro N° 188.540
Síndrome
de Klinefelter
Es la presencia de un cromosoma X extra en un
hombre.
Causas
La mayoría de las personas tienen 46
cromosomas. Los cromosomas contienen todos los genes y el ADN, los pilares
fundamentales del cuerpo. Dos cromosomas sexuales determinan si usted se
convertirá en niño o en niña. Las mujeres normalmente tienen dos cromosomas
sexuales XX, mientras que los hombres normalmente tienen un cromosoma X y un
cromosoma Y.
El síndrome de Klinefelter se presenta
cuando un niño varón nace con al menos un cromosoma X extra. Por lo regular,
esto ocurre debido a un cromosoma X adicional. Esto se escribiría como XXY.
El síndrome de Klinefelter se presenta en
aproximadamente uno de cada 500 a 1,000 bebés varones. Las mujeres que resultan
embarazadas después de los 35 años tienen una probabilidad ligeramente mayor de
tener un niño con este síndrome que las mujeres más jóvenes.
Síntomas
Proporciones corporales anormales (piernas largas, tronco corto, hombro
igual al tamaño de la cadera)
Agrandamiento anormal de las mamas (ginecomastia)
Infertilidad
Problemas sexuales
Vello púbico, axilar y facial menor a la cantidad normal
Testículos pequeños y firmes
Estatura alta
Pruebas y exámenes
El síndrome de Klinefelter primero se puede
diagnosticar cuando un hombre va al médico a consultar debido a la
infertilidad, ya que ésta el síntoma más común.
Se pueden realizar los siguientes exámenes:
Cariotipado
Conteo de semen
Se harán exámenes de sangre para verificar
los niveles hormonales incluyendo:
Estradiol, un tipo de estrógeno
Hormona foliculoestimulante
Hormona luteinizante
Testosterona
Tratamiento
Se puede prescribir la terapia con testosterona
que puede ayudar a:
Promover el crecimiento de vello corporal
Mejorar la apariencia de los músculos
Mejorar la concentración
Mejorar la autoestima y el estado de ánimo
Mejorar la energía y el impulso sexual
Mejorar la fuerza
La mayoría de los hombres con este síndrome
no son capaces de embarazar a una mujer; sin embargo, un especialista en
infertilidad puede ayudarlos. Un médico especial llamado endocrinólogo también
puede ser útil.
Posibles complicaciones
El agrandamiento de los dientes con un
adelgazamiento de la superficie, denominado taurodontismo, es muy común en el
síndrome de Klinefelter y se puede observar en radiografías dentales.
Este síndrome también incrementa el riesgo
de:
Trastorno de hiperactividad y déficit de atención
Trastornos autoinmunitarios como el lupus, la artritis reumatoidea y el
síndrome de Sjögren
Cáncer de mama en hombres
Depresión
Dificultades de aprendizaje, incluso dislexia, que afecta la lectura
Un raro tipo de tumor llamado de células germinativas extragonadales
Enfermedad pulmonar
Osteoporosis
Venas varicosas
Cuándo contactar a un profesional médico
Solicite una cita con el médico si su hijo
no desarrolla características sexuales secundarias en la pubertad. Esto incluye
crecimiento de vello facial y engrosamiento de la voz.
Un asesor en genética puede brindarle
información acerca de esta afección y enviarlo a grupos de apoyo en su área.
Nombres alternativos
Síndrome 47 X-X-Y
Referencias
MedlinePlus http://www.nlm.nih.gov/medlineplus/spanish/ency/article/000382.htm
Bacino CA, Lee B. Cytogenetics. In: Kliegman RM, Behrman RE, Jenson HB,
Stanton BF, eds. Nelson Textbookof Pediatrics.19th ed. Philadelphia, Pa:
Saunders Elsevier; 2011: chap 74.
La
anemia falciforme
La anemia falciforme es una enfermedad de la
sangre de origen hereditario que afecta prioritariamente a las personas que
tienen antepasados de raza negra, aunque también se da en otros grupos étnicos,
incluyendo las personas que tienen antepasados de origen mediterráneo o de
oriente medio.
En EE.UU., hay más de 70.000 personas
afectadas por esta enfermedad. Aproximadamente dos millones de norteamericanos
—uno de cada doce— tiene el rasgo drepanocítico, lo que significa que son
portadores de un único gen de la enfermedad sin manifestarla.
¿Qué es la anemia falciforme?
La anemia falciforme, también conocida como
anemia drepanocítica o drepanocitosis, es un trastorno sanguíneo que afecta a
la hemoglobina, la proteína que contienen los glóbulos rojos y que ayuda a
transportar el oxígeno por todo el cuerpo.
La anemia falciforme ocurre cuando una
persona hereda dos genes anómalos (uno de cada progenitor), lo que determina
que sus glóbulos rojos tengan una forma anómala. En vez de ser flexibles y en
forma de disco, son rígidos y curvos, adoptando la forma de una antigua
herramienta de labranza denominada hoz —de donde proviene el nombre de la
enfermedad (falciforme significa relativo a la hoz)-, parecida a una luna en
cuarto creciente.
Los glóbulos rojos que contienen la
hemoglobina normal (hemoglobina A o HbA) se desplazan fácilmente por el
torrente sanguíneo, repartiendo oxígeno a todas las células del cuerpo. Los
glóbulos rojos normales tienen forma de disco o rosquilla con el centro cóncavo
y son blandos y flexibles, lo que les permite pasar incluso a través de vasos
sanguíneos muy estrechos.
La anemia falciforme tiene lugar cuando se
produce una forma anómala de hemoglobina (HbS). Las moléculas de HbS tienden a
amontonarse, convirtiendo los glóbulos rojos en unas células pegajosas, rígidas
y más frágiles y haciéndoles adoptar una forma similar a la de una hoz.
Los glóbulos rojos que contienen HbS pueden
ir alternando entre la forma normal y la forma anómala hasta que a la larga
acaban adoptando permanentemente la forma anómala. Entonces, en vez de
desplazarse fácilmente por el torrente sanguíneo, pueden obstruir vasos
sanguíneos, impidiendo que los tejidos corporales reciban el oxígeno que
necesitan para funcionar y mantenerse sanos.
A diferencia de los glóbulos rojos normales,
que permanecen aproximadamente 4 meses en el torrente sanguíneo, los glóbulos
rojos anómalos, más frágiles, se descomponen al cabo de 10 a 20 días, lo que
suele provocar anemia. La anemia tiene lugar cuando la cantidad de glóbulos
rojos que hay en el cuerpo (o la cantidad de hemoglobina) está por debajo de lo
normal. Las personas que padecen anemia suelen encontrase débiles, se cansan
con facilidad y pueden tener aspecto de estar “hechas polvo”.
Las personas con anemia falciforme también
pueden tener complicaciones provocadas por los problemas circulatorios, así
como dificultades para luchar contra las infecciones. Por ejemplo, tienen un
riesgo incrementado de padecer determinadas infecciones, así como apoplejía y
un trastorno denominado síndrome torácico agudo, provocado por la inflamación
del tejido pulmonar a consecuencia de los glóbulos rojos que quedan atrapados
en los pulmones.
La anemia falciforme no es contagiosa, de
modo que no te la puede contagiar nadie ni tú se la puedes contagiar a nadie
como si se tratara de un catarro u otra infección. Las personas con anemia
falciforme han heredado dos genes de la drepanocitosis, uno de cada uno de sus
progenitores. Si un niño hereda el rasgo drepanocítico solamente de uno de sus
progenitores, no desarrollará la enfermedad, pero será portador del rasgo. Las
personas que son portadoras de ese rasgo no tienen anemia falciforme ni ningún
síntoma de la enfermedad, pero pueden transmitir el gen de la drepanocitosis a
su descendencia.
Puesto que la gente que solo hereda un gen de
la drepanocitosis nunca llega a desarrollar la enfermedad pero la puede
transmitir a su descendencia, es posible que no sepa que es portadora de ese
gen. Por eso se recomienda a aquellos jóvenes que tengan dudas sobre si son
portadores del gen defectuoso que pidan a sus médicos que les haga una prueba para
dilucidarlo. El Instituto Nacional de la Salud (NIH por sus siglas en inglés)
recomiendan hacer pruebas de cribado a todos los recién nacidos para saber si
padecen anemia falciforme y actualmente estas pruebas son obligatorias en los
recién nacidos de prácticamente todos los estados de EE.UU. Esto permite que
los lactantes que padecen la enfermedad reciban ni bien nacen los cuidados y
tratamientos que necesitan.
Signos y síntomas
Las personas que tienen anemia falciforme
pueden desarrollar ictericia, un trastorno provocado por la elevada tasa de
descomposición de los glóbulos rojos que se caracteriza porque la piel y el
blanco de los ojos adquieren una tonalidad amarillenta.
Las personas con anemia falciforme también
pueden presentar episodios agudos de fuerte dolor en el pecho, el estómago, los
brazos, las piernas y otras partes del cuerpo. Esto se debe a que los glóbulos
rojos anómalos obstruyen el riego sanguíneo en esas partes del cuerpo. Estar
muy cansado y tener problemas para hacer frente a las infecciones también son
síntomas frecuentes entre los jóvenes con anemia falciforme, que pueden tener
trastornos de crecimiento.
Los episodios de dolor, también denominados
crisis de dolor, varían en intensidad, frecuencia y duración. Mientras que
algunas personas tienen un solo episodio de dolor al año, otras experimentan
este tipo de episodios mucho más a menudo. Estos episodios pueden ser de corta
duración o pueden durar horas, días e incluso semanas. El dolor puede afectar a
cualquier órgano o tejido corporal, por ejemplo, los brazos, las piernas, las
caderas y los hombros. Cuando una persona con anemia falciforme tiene un
síndrome torácico agudo, puede presentar fuerte dolor torácico y abdominal,
fiebre, tos y dificultades para respirar.
¿Qué hacen los médicos?
Para diagnosticar la anemia falciforme, los
médicos utilizan una prueba especial para analizar la sangre denominada
electroforesis de la hemoglobina a fin de detectar HbS en la sangre de sus
pacientes.
La anemia falciforme no tiene curación y
algunas personas se ponen tan enfermas que pueden llegar a fallecer a
consecuencia de la enfermedad (aunque la mayoría de jóvenes afectados por esta
enfermedad no mueren). No obstante, existen tratamientos que ayudan a prevenir
las complicaciones de la anemia falciforme. A los jóvenes con anemia falciforme
les suelen recetar ácido fólico, una vitamina que ayuda al cuerpo a producir
nuevos glóbulos rojos. Los analgésicos (medicamentos para aliviar el dolor)
ayudan a aliviar los síntomas en los episodios de dolor. Asimismo, los niños y
jóvenes con anemia falciforme deberían tomar penicilina u otros antibióticos
para ayudar a prevenir las infecciones.
Algunas crisis se pueden tratar en casa con
analgésicos, reposo e ingesta de líquido en abundancia. Pero, si una crisis es
especialmente intensa, se deberá ingresar al paciente en el hospital para que
le administren fluidos por vía intravenosa (IV) y analgésicos más potentes. Los
afectados también pueden requerir oxígeno para aliviar los síntomas durante las
crisis o episodios de síndrome torácico agudo.
Los jóvenes que padecen esta enfermedad
pueden requerir transfusiones de glóbulos rojos sanos para que el oxígeno pueda
llegar más eficazmente a todos los tejidos corporales, y algunos de ellos
necesitan someterse a transfusiones regularmente.
Los científicos están investigando
constantemente para encontrar nuevas formas de ayudar a las personas afectadas
por esta enfermedad. Varios tratamientos nuevos, como el fármaco hidroxiurea,
han ayudado a reducir las crisis de dolor y los episodios de síndrome torácico
agudo en adultos.
Fuente: The Nemours Foundation.
http://kidshealth.org/teen/en_espanol/enfermedades/sickle_cell_anemia_esp.html#
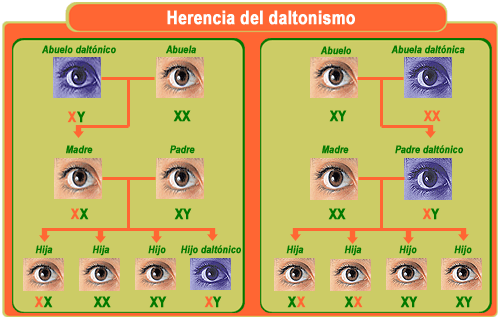
Daltonismo
Aproximadamente uno de cada diez hombres
presenta alguna de las formas de daltonismo, una incapacidad visual para
distinguir ciertos colores, que tiene un origen genético ligado al cromosoma X.
¿Cómo vemos los colores?
El ojo humano está formado por distintas
estructuras que interaccionan para permitir la recepción de los estímulos
visuales.
Uno de los componentes del ojo es la retina,
que se encuentra situada en la parte posterior del ojo y se encarga de
“proyectar” las imágenes (estímulos) que le llegan del exterior. En esta parte
del ojo se encuentran las únicas células capaces de captar el color y la luz,
son los denominados conos y bastones.
Existen tres tipos de conos, cada uno de los
cuales puede captar las longitudes de onda equivalentes al rojo, azul y verde,
gracias a su contenido en pigmentos (moléculas que captan la luz a diferentes
longitudes de onda –lo que equivale a los distintos colores-). Mediante las
diferentes intensidades captadas por los tres tipos de conos, es posible
distinguir todos los colores que forman parte del espectro de luz visible.
¿Qué es el daltonismo?
Esta alteración debe su nombre al científico
inglés John Dalton, que fue el primer caso descrito de daltonismo. El
daltonismo es una incapacidad visual que impide distinguir ciertos colores.
Esto se debe a la falta o al mal funcionamiento de uno o más de un tipo de
cono.
Existen múltiples variantes de este
trastorno; en realidad, podría decirse que ningún daltónico ve exactamente
igual que otro. Haciendo una clasificación a groso modo se pueden distinguir
tres tipos de daltonismo:
Dicromatismo
La forma más común es el dicromatismo, que
afecta a los conos encargados de captar el rojo o el verde. Al faltar uno de
los tipos celulares, el otro se encargará de recoger los estímulos que
corresponderían al primero; así, en muchos casos los dos estímulos entrantes
serán percibidos como un mismo color. Las personas que presentan este tipo de
daltonismo tienen dificultad para establecer la diferencia entre los colores
rojo y verde.
Otra variante de este trastorno es la falta
de los conos encargados de captar los tonos azules; en este caso, los
individuos confundirán con frecuencia los colores azul y amarillo. Estos tipos
de daltonismo son denominados dicromatismos, puesto que el individuo posee solo
dos tipos de conos.
Tricromatismo anómalo: Otra de las formas de
daltonismo que tiene efectos similares, aunque más leves, que los dos casos
anteriores. En este caso, el individuo presenta los tres tipos de conos, pero
existe alguna deficiencia en los mismos que impide un funcionamiento totalmente
normal.
Acromatopsia: Por último, el caso más grave
de daltonismo es la denominada acromatopsia, a consecuencia de la cual el
individuo que la padece aprecia únicamente diferencias en la escala de grises.
Causas del daltonismo
Esta alteración tiene un origen genético; se
trata de un trastorno de herencia ligada al sexo, es decir, el gen afectado se
encuentra en uno de los cromosomas sexuales (el ser humano tiene 46 pares de
cromosomas de los cuales 22 pares son autosómicos y un par es sexual).
En este caso se trata de un gen recesivo
ligado al cromosoma X; esto quiere decir que todos los hombres que hereden un
cromosoma X con el gen defectuoso padecerán el trastorno y que las mujeres, en
cambio, solo lo padecerán en caso de que ambos cromosomas presenten el gen (lo cual
es bastante improbable, ya que requeriría que los dos progenitores portasen
dicho gen). Esto explica que los casos de daltonismo en mujeres sean poco
frecuentes, mientras que aproximadamente uno de cada diez hombres presenta
alguna de las formas de daltonismo.
Diagnóstico del daltonismo
En la mayoría de los casos de daltonismo,
excepto en los más leves, el paciente y las personas de su entorno comienzan a
detectar ciertas anormalidades visuales durante el desempeño de las actividades
diarias, escolares, etc. Otras muchas veces los individuos descubren por
casualidad que padecen este tipo de trastorno.
Existe diferentes pruebas y test para el
diagnóstico del daltonismo y el grado de la alteración. Así, las manchas de
Ishihara y Stilling consisten en láminas en las que aparecen puntos de colores
primarios dispuestos formando números o formas, sobre un fondo de puntos de
colores similares, de modo que una persona con una capacidad visual normal
podrá distinguirlos, mientras que un daltónico no. Además de estas pruebas, es
recomendable realizar un reconocimiento oftalmológico confirmativo para
asegurar el diagnóstico.
Test de Ishihara
El test de Ishihara, que se llama así en
honor a su creador, el doctor Shinobu Ishihara, consta de 38 cartas que
muestran un círculo con puntos de colores de diversos tamaños. Los puntos
forman un número que es visible para las personas que no padecen ningún defecto
visual, pero aquellos con algún grado de daltonismo ven números distintos o,
directamente, no son capaces de ver ningún número. Aunque la prueba completa
incluye 38 discos, con que el paciente observe unas cuantas cartas es
suficiente para detectar la existencia de un defecto visual. A continuación se
presentan unas cuantas figuras correspondientes a las cartas de Ishihara.
Escrito por Natalia Dudzinska Camarero,
bióloga disponible en:
http://www.webconsultas.com/daltonismo/daltonismo-2632
Albinismo
El albinismo es un defecto hereditario que
altera la producción y el metabolismo de la melanina, lo que se traduce en una
ausencia total o parcial de este pigmento que es responsable del color de la
piel, cabello y ojos.
El albinismo es un defecto hereditario que
altera la producción y el metabolismo de la melanina, lo que se traduce en una
ausencia total o parcial de este pigmento producido por unas células llamadas
melanocitos y que es responsable del color de la piel, cabello y ojos.
Los afectados por el albinismo son llamados
albinos y carecen de pigmentos en el cabello, la piel o el iris del ojo. La
falta de pigmentación de la piel hace que los albinos sean más susceptibles a
las quemaduras solares y al cáncer de piel. Y es que el papel fisiológico
fundamental de la melanina es proteger la piel de los efectos nocivos de la luz
ultravioleta, es por ello que las personas que habitan cerca de los trópicos
son más oscuras, ya que su exposición al sol es mayor, en comparación con las
personas que viven en climas templados.
El patrón de herencia del albinismo es de
tipo autosómico recesivo, lo cual significa que la probabilidad de que el
trastorno se transmita de una generación a otra es baja. Las estadísticas
muestran que alrededor de una de cada 17.000 personas sufren de algún tipo de
albinismo.
La persecución a los albinos
El albinismo provoca rechazo social y, lo que
es peor, los albinos o, mejor dicho, algunas partes de su cuerpo, como la
sangre, los huesos, o los órganos, se consideran en algunos países africanos amuletos
contra el ‘mal de ojo’.
Así, a los problemas de salud que les puede
ocasionar su condición de albinos –la radiación solar daña su piel y sus ojos,
que son mucho más sensibles que los del resto de la población, y sin las
medidas de prevención y el tratamiento adecuados pueden desarrollar cáncer de
piel desde niños–, estas personas añaden un estigma social a causa de su
aspecto que, si además han nacido en determinados países, les puede llegar a
costar la vida. En Tanzania, por ejemplo, hay una creencia popular según la
cual preparar y tomar pociones que contengan partes del cuerpo de un albino,
proporciona suerte y riquezas.
El problema se agrava, además, porque el
albinismo en Tanzania tiene una gran incidencia y se estima que afecta a más de
150.000 personas, aunque se desconoce el número exacto. Los albinos son
sometidos a la persecución y el linchamiento, y se ven obligados a ocultarse y
a abandonar sus hogares. Por ello, organismos como la Cruz Roja y algunas
ONG’s, como la Asociación para la Integración y Progreso de las Culturas –
Pandora (AIPC Pandora), han puesto en marcha proyectos para proteger a estos
fugitivos involuntarios, y en 2010 se creó ‘Kabanga School’, en el norte de
Tanzania, un centro donde se facilita a los albinos el acceso a profesionales
médicos que evalúen y traten sus problemas físicos y establezcan medidas para
prevenir la ceguera, y se les ofrecen actividades educativas, culturales y de
ocio, especialmente a los menores.
Dermatólogos españoles, que acuden como
voluntarios a Tanzania para enseñar a los médicos locales cómo manejar las
lesiones de la piel de los albinos, han podido comprobar la discriminación de
la que son objeto estas personas, y se han asombrado de la capacidad de
resistencia ante el dolor de los niños afectados por este trastorno. Según los
médicos, estos niños son tímidos y desconfiados, y rara vez sonríen –algo
natural teniendo en cuenta el rechazo social y el riesgo que corren de ser
asesinados–, pero no lloran ni se quejan a pesar de las lesiones que sufren a
consecuencia de su blanquísima piel, incapaz de defenderse frente a un sol que,
en África, brilla con intensidad casi todos los días del año.
Causas y tipos de albinismo
Para entender las causas del abinismo tenemos
que comprender que el proceso bioquímico por el cual se produce la melanina se
llama melanogénesis, y en este participan numerosas enzimas, entre ellas una de
las más importantes es la enzima tirosinasa que convierte la tirosina (un
aminoácido) en una sustancia llamada dopa y posteriormente en dopaquinona, y es
además la responsable del incremento en la pigmentación del iris, que se
produce en los bebés durante los primeros seis meses de vida.
Si un individuo presenta una mutación en el
gen de la tirosinasa, que se encuentra ubicado en el brazo largo del cromosoma
11, esto puede traducirse en una ausencia completa o parcial de la enzima o
bien resultar en una enzima no funcional, lo que trae como consecuencia un
déficit en la producción del pigmento melanina y por ende causar albinismo.
En cualquier caso si la persona presenta la
misma mutación en el gen heredado del padre y en el gen heredado de la madre se
dice que es homocigoto, si la mutación es diferente se denomina heterocigoto.
Tipos de albinismo
En realidad se puede hablar de varios tipos
de albinismo en función de las siguientes cuestiones:
Según la afectación o no de la piel
Albinismo óculo-cutáneo: es el más frecuente y afecta la piel, los ojos
y el cabello.
Albinismo ocular: que afecta únicamente los ojos.
Según el defecto de la enzima implicada en la
melanogénesis
Albinismo óculo-cutáneo tipo 1: se debe a un defecto genético enzima la
tirosinasa. Hay dos subtipos de esta forma de albinismo
Subtipo 1A: es la forma más frecuente y severa de albinismo, la enzima
yirosinasa es inactiva y no se produce melanina, lo que conduce a individuos
con el pelo blanco, piel muy clara y muchas alteraciones visuales.
Subtipo 1B: la enzima es mínimamente activa y se produce una pequeña
cantidad de melanina, lo que conduce a que el cabello pueda oscurecerse al
rubio, de color marrón claro amarillo o incluso naranja, así como más pigmento
en la piel y una tez más oscura pero blanca.
Albinismo óculo-cutáneo tipo 2: afectación del gen P que resulta en una
proteína P defectuosa. Esta proteína colabora en la función de la enzima
tirosinasa, por lo tanto, los individuos con este tipo de albinismo producen
cantidades mínimas de melanina y puede tener color de cabello que van desde el
rubio muy claro a marrón.
Albinismo óculo-cutáneo tipo 3: rara vez se ha descrito y resulta de un
defecto genético en TYRP1, una proteína relacionada con la tirosinasa. Los
individuos con albinismo tipo 3 pueden tener pigmento sustancial.
Albinismo óculo-cutáneo tipo 4: resulta de un defecto genético en la
proteína SLC45A2, que ayuda a la enzima tirosinasa para su función. Los
individuos con albinismo tipo 4 producen cantidades mínimas de melanina similar
a las personas con albinismo tipo 2.
Albinismo óculo-cutáneo con manifestaciones sistémicas: es el también llamado
Síndrome de Hermansky-Pudlak, en el cual existe una anormalidad en la formación
de dos tipos vesículas intracelulares: los melanosomas (que contienen la
melanina) en los melanocitos y los cuerpos densos de las plaquetas. Esto trae
como consecuencia que los pacientes, además, de presentar albinismo, tengan una
predisposición al sangrado de vasos sanguíneos pequeños como los cutáneos o los
cerebrales. Este trastorno es más común en Suiza y Puerto Rico. Cuando se
realiza una biopsia de piel a estos individuos se observan melanosomas
gigantes, llamados macromelanosomas. Aproximadamente el 50% de los pacientes
con este síndrome pueden presentar con la edad problemas pulmonares (enfermedad
pulmonar intersticial), problemas inflamatorios crónicos en el colon (colitis
granulomatosa), problemas renales (insuficiencia renal), o cardiacos
(miocardiopatía).
Signos y síntomas del albinismo
La severidad de los síntomas y problemas
clínicos asociados al albinismo varía de un individuo a otro, dependiendo del
tipo de defecto genético que padezcan. Sin embargo, las patologías asociadas a
esta condición se pueden agrupar en dos grandes tipos de problemas: de la piel
y de la visión.
Problemas de la piel en albinos
Aunque la mayoría de las personas con
albinismo son de tez blanca pálida, el color de la piel o del cabello no son
por si solos signos que indiquen un diagnóstico fehaciente de albinismo.
Entre los síntomas del albinismo tenemos que
la escasa pigmentación de la piel produce en estos individuos una alta
sensibilidad a las quemaduras producidas por la exposición al sol, así como una
elevada susceptibilidad a padecer algún tipo de cáncer de piel. Así mismo
tienen riesgo de presentar un engrosamiento patológico de la piel que se llama
queratosis, o coloración rojiza de la piel, eritematosis.
Las personas con albinismo deben tomar
precauciones para evitar daños en la piel causados por el sol, tales como el
uso de lociones con protección solar, sombreros, camisas de manga larga así
como pantalones largos.
Problemas de la visión en albinos
Las personas con albinismo siempre tienen
problemas de la visión, por lo general no corregibles con gafas. El grado de
deterioro de la vista varía con los diferentes tipos de albinismo y algunos
pueden llegar a ser "legalmente ciegos", sin embargo la mayoría puede
usar su visión para muchas tareas, incluyendo la lectura, por lo que no
utilizan el lenguaje Braille. Algunos albinos tienen la visión suficiente para
conducir un coche. Los problemas de visión son el resultado de un desarrollo
anormal de la retina y patrones anormales de conexiones nerviosas entre el ojo
y el cerebro. Es la presencia de estos problemas de los ojos lo que define el
diagnóstico de albinismo. Entre los problemas visuales más frecuentes en los
albinos podemos encontrar:
Estrabismo: desequilibrio de los músculos de los ojos, "ojos
cruzados o bizcos" (esotropía), "ojo perezoso" o un ojo que se
desvía hacia fuera (exotropía).
Fotofobia: sensibilidad aumentada a la luz brillante y el resplandor.
Astigmatismo: error de refracción causado por una imperfección en la
curvatura de la córnea, lo que impide el enfoque claro de los objetos cercanos.
Hipoplasia foveal: falta de desarrollo de la fóvea del ojo y del nervio
óptico.
Trayectoria anormal del nervio óptico: las señales nerviosas que viajan
desde la retina al cerebro no siguen las rutas habituales.
Hipopigmentación del iris: en los pacientes albinos, la zona de color en
el centro del ojo tiene poco o ningún pigmento para filtrar la luz difusa que
entra en el mismo. Normalmente, la luz entra en el ojo sólo a través de la
pupila, la abertura oscura en el centro del iris, pero en el albinismo la luz
puede pasar a través del iris también, esto se llama transiluminación del iris.
Nistagmo: en la primera infancia, el nistagmo (movimiento involuntario e
incontrolable de los ojos) tiene una gran amplitud y baja frecuencia (forma de
onda triangular), un patrón que a veces hace que los padres piensen que su niño
es incapaz de fijar objetos. Con la edad, el nistagmo eventualmente madura en
una forma pendular; virtualmente cualquier tipo de movimiento es posible,
incluyendo vertical y rotatorio.
Diagnóstico y tratamiento del albinismo
El albinismo es una condición que se puede
diagnosticar simplemente por la observación de la ausencia parcial o total de
la pigmentación de la piel, cabello y ojos.
Dentro del conjunto de pruebas que se pueden
realizar para verificar un diagnóstico de albinismo tenemos: pruebas genéticas
para detectar las mutaciones causantes del albinismo; biopsias de piel en la
que se puede observar ausencia del pigmento melanina, o el electrorretinograma.
Este test es una prueba no invasiva que consiste en la colocación de un par de
pequeños electrodos de registro, uno que se coloca en el párpado del ojo y otro
sobre la córnea, se procede a presentar estímulos visuales al paciente como
destellos de flash o la imagen de una rejilla. La respuesta de la retina a
estos estímulos luminosos se detecta como impulsos eléctricos y se registra una
gráfica que en los pacientes albinos está por encima de lo normal.
Pronóstico y tratamiento del albinismo
Las personas que tienen la condición de
albinos pueden vivir una vida normal, sin embargo, las vidas de las personas
con Síndrome de Hermansky-Pudlak se puede acortar por la enfermedad pulmonar o
cardiaca.
En general los individuos con albinismo deben
ser valorados desde el nacimiento por un equipo multidisciplinario de
especialistas constituido por pediatras, oftalmólogos, dermatólogos, psicólogos
y asistentes sociales, ya que el albinismo puede causar problemas de adaptación
social, pues la condición de estos pacientes les confiere un aspecto diferente
al de sus familiares, compañeros y otros miembros de su grupo étnico.
No existe cura para la condición de albino,
solo se pueden realizar tratamientos dirigidos a prevenir el envejecimiento
prematuro y el cáncer de piel.
Para el tratamiento del albinismo a nivel
visual se puede realizar cirugías para corregir el estrabismo, y en el caso de
la esotropía o la extropía, la cirugía puede ayudar a la visión mediante la
ampliación del campo visual (el área que los ojos pueden ver mientras se mira
en un punto). Pero en su mayor parte, el tratamiento de las afecciones del ojo
consta de rehabilitación visual, uso de gafas bifocales, utilización de gafas
de sol para disminuir la fotofobia, incluso algunos recurren a lupas de mano o
pequeños telescopios especiales y otros prefieren usar la magnificación de
pantalla en los ordenadores.
Escrito
por Marta Talise, licenciada en medicina y análisis clínico
http://www.webconsultas.com/salud-al-dia/albinismo/albinismo-8937
La
Hemofilia
La hemofilia es un problema hemorrágico. Las
personas con hemofilia no sangran más rápido que lo normal, pero pueden sangrar
durante un período más prolongado. Su sangre no contiene una cantidad
suficiente de factor de coagulación. El factor de coagulación es una proteína
en la sangre que controla el sangrado.
La hemofilia es bastante infrecuente.
Aproximadamente 1 en 10.000 personas nace con ella.
Tipos de hemofilia
El tipo más común de hemofilia se llama
hemofilia A. Esto quiere decir que la persona no tiene suficiente factor VIII
(factor ocho).
Un tipo menos común es la denominada
hemofilia B. Esta persona no tiene suficiente factor IX (factor nueve). El
resultado es el mismo para la hemofilia A y B, o sea, un sangrado por un tiempo
mayor que el normal.
La hemofilia se hereda; esto quiere decir que
se transmite a través de los genes de la madre y del padre. Los genes portan
mensajes acerca de la manera en que las células del organismo se desarrollarán
a medida que un bebé va creciendo hasta hacerse adulto. Los genes determinan,
por ejemplo, el color de pelo y de ojos de una persona.
A veces, la hemofilia puede ocurrir sin que
haya antecedentes familiares de la enfermedad. Ésta se denomina hemofilia
esporádica. Alrededor del 30% de las personas con hemofilia no la contrajeron
por medio de los genes de sus progenitores. Fue causada por un cambio en sus
propios genes.
¿Cómo se hereda la hemofilia?
El gene de la hemofilia pasa de uno de los
padres al hijo. Los genes de la hemofilia A y B se encuentran en el cromosoma
X. Por eso, la hemofilia se describe como ‘ligada al cromosoma X’. Cuando el
padre tiene hemofilia pero la madre no, ninguno de los hijos varón tendrá
hemofilia. Todas las hijas portarán el gen de la hemofilia.
Las mujeres que tienen el gen de la hemofilia
son llamadas portadoras. A veces muestran
signos de la hemofilia, y pueden transmitirla a sus descendientes. Para
cada uno de sus descendientes, hay un 50% de probabilidad de que si es varón
tenga hemofilia y un 50% de probabilidad de que si es mujer sea portadora del
gen.
Una mujer únicamente puede tener hemofilia si
su padre tiene hemofilia y su madre es portadora. Esto es muy infrecuente.
Grados de severidad
La severidad describe cuán serio es el
problema. El nivel de la severidad depende de la cantidad de factor de coagulación que falta en la sangre de la
persona.
Muchas personas con hemofilia severa padecen
de hemorragias frecuentes en músculos o articulaciones. Sin tratamiento
preventivo, pueden sangrar una o dos veces por semana. La hemorragia es con
frecuencia espontánea, lo que quiere decir que ocurre sin causa aparente.
Las personas con hemofilia moderada padecen
hemorragias con menos frecuencia, sobre una vez al mes. Pueden sangrar durante
mucho tiempo tras una cirugía, una lesión seria, o procedimientos
odontológicos. Rara vez, sangran sin que haya un motivo claro.
Las personas con hemofilia leve por lo
general sólo sufren de hemorragias a consecuencia de cirugías o lesiones
graves. Podrían nunca llegar a tener un problema de sangrado.
Síntomas y diagnóstico
Los signos de la hemofilia A y B son los
mismos:
Hematomas
extensos
Sangrado
dentro de los músculos y las articulaciones
Sangrado
espontáneo (sangrado repentino dentro del cuerpo sin que haya un motivo claro)
Sangrado
durante mucho tiempo tras cortarse, sacarse una muela o someterse a una cirugía
Sangrado
durante mucho tiempo tras sufrir un accidente, particularmente luego de una lesión en la cabeza
El sangrado dentro de una articulación o un
músculo provoca:
Dolor
o “una sensación extraña”
Hinchazón
Dolor
y rigidez
Dificultad
para utilizar una articulación o músculo
¿Cuáles son los lugares más frecuentes de
sangrado?
Las personas con hemofilia pueden tener
hemorragias internas o externas. La mayoría de las hemorragias tienen lugar en las
articulaciones o músculos: particularmente en las rodillas, los codos, y los
tobillos, así como los músculos del brazo superior y del antebrazo, el músculo
de psoas, del muslo, y de la pantorrilla.
Si hay sangrado repetidas veces en una misma
articulación, dicha articulación puede dañarse y doler.
Las hemorragias repetidas pueden causar otros
problemas de salud, como artritis. Esto puede provocar dificultad para caminar
o para realizar actividades sencillas. Sin embargo, las articulaciones de las
manos generalmente no están afectadas en la hemofilia (a diferencia de lo que
ocurre en algunos tipos de artritis).
¿Cómo se diagnostica la hemofilia?
La hemofilia se diagnostica tomando una
muestra de sangre y midiendo el grado de actividad del factor. La hemofilia A
se diagnostica haciendo pruebas del grado de actividad de coagulación del
factor VIII. La hemofilia B se diagnostica midiendo el grado de actividad del
factor IX.
Si la madre es portadora, pueden hacerse
pruebas antes del nacimiento del bebé. El diagnóstico prenatal puede realizarse
entre la 9a y la 11a semana del embarazo mediante una muestra del velo
coriónico o de una muestra de sangre fetal en una etapa posterior (18 semanas o
más de embarazo).
Estas pruebas pueden realizarse en un centro
de tratamiento de hemofilia. La página Internet de la FMH cuenta con una lista
de centros de tratamiento en todo el mundo. Haga clic en Pasaporte para hacer
una búsqueda en el directorio.
Tratamiento
Hoy día, el tratamiento de la hemofilia es
muy efectivo. Se inyecta el factor de coagulación faltante al torrente
sanguíneo utilizando una aguja. El sangrado se detiene cuando una cantidad
suficiente de factor de coagulación llega al sitio que está sangrando.
Los sangrados deben tratarse rápidamente. El
tratamiento precoz le ayudará a disminuir el dolor y el daño a las
articulaciones, músculos y órganos. Si el sangrado es tratado prontamente, se
necesitará una menor cantidad de factor coagulante para detener la hemorragia.
Con el tratamiento adecuado, las personas con
hemofilia pueden llevar vidas perfectamente saludables. Sin él, muchas morirán
jóvenes. Lamentablemente, sólo cerca del 25 por ciento de las 400 mil personas
que se calcula padecen hemofilia recibe tratamiento adecuado. La FMH procura
mejorar estas estadísticas.
Opciones de tratamiento
Los concentrados de factor constituyen el
tratamiento preferido para la hemofilia. Pueden fabricarse a partir de sangre
humana (conocidos como productos hemoderivados) o utilizando células
genéticamente diseñadas que portan un gene de factor humano (llamados productos
recombinantes). Los concentrados de factor se elaboran en sofisticadas
instalaciones de fabricación. Todos los concentrados de factor de preparación comercial
son tratados para eliminar o inactivar virus transportados por la sangre.
El crioprecipitado es derivado de la sangre y
contiene una moderada concentración de factor VIII (pero no de factor IX). Es
eficaz para hemorragias articulares y musculares, pero menos seguro que los
concentrados en términos de contaminación viral; además, su almacenamiento y
administración son más difíciles. El crioprecipitado puede elaborarse en
instalaciones locales de recolección de sangre.
El plasma fresco congelado (PFC), del que se
han eliminado los glóbulos rojos dejando las proteínas sanguíneas y los
factores de coagulación es menos eficaz que el crioprecipitado para el
tratamiento de la hemofilia A porque el factor VIII está menos concentrado. Es
necesario transfundir grandes volúmenes de plasma. Esto puede causar una
sobrecarga circulatoria.
Las personas con hemofilia A leve a veces
utilizan desmopresina (también llamada DDAVP), una hormona sintética que
estimula la liberación de factor VIII, para tratar sangrados menores.
Fuente:
http://www.wfh.org/es/page.aspx?pid=932
La
Polidactilia
La polidactilia (del griego «poly»=«mucho» y
«daktylos»=«dedo») es un trastorno genético donde un humano nace con más dedos
en la mano o en el pie de los que le corresponde (por lo regular un dedo más).
Se detecta en el momento del nacimiento o
veces antes por medio del ultrasonido. A estos dedos se les llaman
“dedos extra” o “dedos supernumerarios”.
La polidactilia afecta a la mano o al pie en
diferentes formas pero no es una malformación que afecte a la salud de la
persona aunque, en algunos casos, puede estar asociada a una condición genética
más seria en la que pueden existir otras malformaciones físicas o mentales.
Este problema afecta a 1 de cada 1000 bebés
nacidos vivos y es más frecuente en la raza negra que en la blanca. No hay
diferencia con relación al sexo.
Causas
Durante el desarrollo normal de la mano
durante el embarazo, al principio tiene forma de paleta que eventualmente se
separará formando los dedos. Algunas veces hay un error en este proceso y los
dedos no se separan bien y el resultado es dos dedos pegados entre sí, a esto
se le llama “sindactilia”, pero otras veces se forman dedos extras y entonces
se da la “polidactilia”. Puede ser que se presenten los dos trastornos
combinados.
El desarrollo embriológico del miembro superior
se realiza desde las primeras etapas del embarazo, presentando una completa
diferenciación a la 7ª semana de gestación. Por ello, cuando la madre ha
confirmado su embarazo, la lesión ya está establecida.
¿Por qué el bebé tiene esta malformación? En
realidad no es debido a NADA que la madre haya hecho durante el embarazo, este
problema puede ser causado por un trastorno en los cromosomas, pero también
puede presentarse naturalmente por un solo gen que puede causar algunas
variaciones sin que haya ningún otro síntoma o enfermedad y simplemente sucede
sin que haya ninguna explicación.
En el 82% de los casos, la polidactilia es un
defecto aislado y su asociación con otras malformaciones es mínima. De todas
maneras, es aconsejable que el médico busque si hay otras malformaciones
asociadas. La polidactilia también se puede transmitir de padres a hijos.
Diagnóstico
El médico hará el diagnóstico basándose en
los antecedentes familiares (preguntará si hay alguien más en la familia
cercana con este problema) y en la historia clínica. Hará también un examen
físico del bebé para descartar la presencia de otras alteraciones que hagan
sospechar de algún trastorno genético.
Algunas de las preguntas de la historia
clínica pueden ser:
¿Algunos otros miembros de la familia han nacido con dedos extras en las
manos o en los pies?
¿Hay antecedentes familiares de cualquier trastorno ligado a la
polidactilia?
¿Hay algunos otros síntomas o problemas?
El médico pedirá también algunos estudios de
laboratorio:
Estudios cromosómicos: Por medio de esta prueba se puede:
Contar el número de cromosomas
Buscar cambios estructurales en los cromosomas
Radiografías de la mano afectada para determinar la morfología del dedo.
Durante el embarazo, esta afección se puede
diagnosticar con ecografía
Comúnmente todos los seres humanos tienen
cinco dedos en cada mano, que van de adentro hacia afuera con la palma hacia
arriba de la siguiente manera: pulgar, índice, medio, anular y meñique. Todos
los dedos de las manos están formados por tres huesos pequeños llamados
falanges (primera falange, segunda falange, en medio, y tercera falange que
forma la punta del dedo).
En la polidactilia los dedos adicionales
pueden ser bien formados e incluso funcionales o, con desarrollo insuficiente y
unidos por un pequeño pedículo; a veces contiene hueso sin articulaciones. El
dedo extra suele ser una bifurcación de un dedo normal y rara vez nace de la
muñeca como los demás dedos.
Cuando un dedo se duplica, generalmente cada
uno de esos dedos son más pequeños que lo normal y es muy común que los dedos
extras se angulen hacia los lados.
Clasificación
La polidactilia puede darse:
En
el primer dedo o pulgar que se encuentra a la altura de un hueso del brazo
llamado radio (llamado preaxial);
Entre cualquiera de los dedos o,
En
el quinto dedo o meñique que está localizado a la altura del cúbito, el otro
hueso del brazo (llamado postaxial).
La duplicación puede afectar a segmentos más
amplios o incluso a toda la extremidad.
Según la ubicación del dedo extra, la
polidactilia se ha clasificado de la siguiente manera:
Preaxial 1: Polidactilia del pulgar o primer ortejo del pie, o también
puede haber implantación en la primera falange del pulgar
Preaxial 2: Pueden existir dos pulgares completos con tres falanges. El
pulgar normal sólo tiene dos falanges.
Preaxial 3: polidactilia del dedo índice, El pulgar extra puede o no
tener tres falanges.
Polidactilia del meñique ó postaxial: polidactilia en el borde de la
mano del lado del hueso cúbito del brazo o peroneo del pie. En éste caso se
pueden dar dos tipos de polidactilia, una es cuando el dedo extra está bien
formado y pegado en la mano; y la segunda, cuando el dedo extra está
pediculado, o sea que no tiene huesos, y sólo cuelga del meñique normal. Esta
última polidactilia es la más frecuente de las dos.
Polidactilia del meñique ó postaxial:
polidactilia en el borde de la mano del lado del hueso cúbito del brazo o
peroneo del pie. Polidactilia del meñique ó postaxial: polidactilia en el borde
de la mano del lado del hueso cúbito del brazo o peroneo del pie.
Duplicación Central: es la presencia de polidactilia en los dedos,
anular, medio o índice, asociándose en forma frecuente sindactilia (Fusión de
piel o huesos de uno o más dedos). Suelen ser bilaterales.
Duplicación Central: es la presencia de polidactilia
en los dedos, anular, medio o índice, asociándose en forma frecuente
sindactilia
Tomando en cuenta las características
clínicas de los dedos supernumerarios y sus repercusiones en los elementos
vecinos, Stelling y Turek la clasifican en:
Tipo I: masa de tejido blando desprovista de
hueso y cartílago.
Tipo
I: masa de tejido blando desprovista de hueso y cartílago.
Tipo II: dedo extra que no está completamente desarrollado.
Tipo III: dedo extra similar al dedo normal adyacente.
Tipo
III: dedo extra similar al dedo normal adyacente.
Tratamiento
El tratamiento es quirúrgico y la cirugía es
programada. Lo ideal es que la extirpación del dedo extra se haga mediante
cirugía plástica.
Cuando el dedo es pequeño y rudimentario,
generalmente se remueven simplemente atando una ligadura alrededor del pedículo
para que lo haga desprenderse más adelante.
Cuando estos dedos son más grandes, se
requiere de una cirugía para extirparlos. La cirugía se debe realizar lo más
pronto posible después de ser detectada, para que la cicatriz sea menor. La
edad óptima de intervención es a partir de los 6 meses, pero cuando el dedo
extra compromete la función de la mano o de los dedos, se debe realizar una cirugía
precoz.
El médico especialista en mano tratará de
lograr que la apariencia y funcionalidad de la mano sean lo mejor posible.
Después de la cirugía es normal proteger la
mano con vendajes durante semanas o meses, dependiendo del tipo de procedimiento
que se haya realizado. En algunas ocasiones es necesario realizar un retoque
cuando el niño crece.
Terapista de mano: En algunas ocasiones un terapista de mano puede
ayudar con algunos problemas que aparecen después de la cirugía aunque algunos
niños se recuperan sin esta ayuda.
Cirugía del pie: La cirugía está indicada
para mejorar la apariencia del pie y el uso del calzado. Generalmente se
realiza cuando el bebé ha cumplido un año de edad para que el efecto en el
desarrollo de la marcha sea mínimo, pero el médico generalmente esperará a que
haya ocurrido la osificación (formación de los huesos) para que pueda hacer una
valoración anatómica adecuada.
Prevención
Los padres que han tenido un hijo con
polidactilia, así como los que tiene antecedentes en la familia, deben de
acudir a consejo genético para determinar el riesgo de que se vuelva a
presentar.
Complicaciones
Imposibilidad de realizar funciones
adecuadamente con la mano (impotencia funcional), sobre todo cuando es el dedo
pulgar el que está de extra.
Complicaciones asociadas: existe la
polidactilia aislada, la cual no se asocia a ningún otro defecto. Pero por
coincidencia puede asociarse a otros, como una cardiopatía o defecto de corazón
congénito.
Lo que sí resulta más frecuente es la
polidactilia con sindactilia (no se desarrolla bien la separación de los dedos
y se quedan dos pegados), la cual es más frecuente en los dedos de los pies.
De cualquier forma es importante buscar otras
manifestaciones y malformaciones ya que la polidactilia puede ser parte de
síndromes.
Pronósticos
En general es favorable excepto cuando el
problema está asociado a otras malformaciones.
En muchos casos, la cirugía da como resultado
una mano muy mejorada pero no totalmente normal en apariencia. Afortunadamente,
a menos de que haya algo particularmente notorio en la mano, lo que la gente
nota más en una persona no es si un dedo es más pequeño que el otro sino más
bien la forma en la que usa su mano.
¿Qué pasa si no se opera?
La polidactilia no tiene ningún riesgo para
la salud y la cirugía generalmente tiene un buen resultado si se hace en los
primeros años de vida cuando el niño tiene tiempo para adaptarse y acomodarse a
los cambios de su mano. Esto es más difícil que suceda cuando la cirugía se
hace después de la infancia.
Probabilidad de que se repita
En algunos casos donde se hereda de padres a
hijos, los afectados tienen un riesgo de transmitirlo a su descendencia del 50
por ciento. El hijo que no presenta la sintomatología no tiene posibilidad de
heredar a su descendencia. Pero la
mayoría de los casos son esporádicos (no hay antecedentes familiares) y no hay
riesgo de repetición.
Referencias
http://www.nlm.nih.gov/medlineplus/spanish/ency/article/003176.htm
http://www.genome.gov/GlossaryS/index.cfm?id=157
http://www.umm.edu/esp_ency/article/003176.htm
http://www.salud.es/polidactilia/sintomas
No hay comentarios:
Publicar un comentario